Analyzing Viral Genome Assembly Using BV-BRC Tools Tutorial¶
Made by Anna Capria with Scribe¶
Overview¶
1. The Viral Genome Assembly Service allows users to assemble viral genomes utilzing IRMA(1)(currently the BV-BRC tool supports Influenza). Once the assembly process has started by clicking the Assemble button, the genome is queued as a “job” for the Assembly Service to process, and will increment the count in the Jobs information box on the bottom right of the page. Once the assembly job has successfully completed, the output file will appear in the workspace.
This service and tutorial are currently in beta- and will be updated to reflect the tools development.
A genome assembly is the sequence produced after chromosomes from the organism have been fragmented, those fragments have been sequenced, and the resulting sequences have been put back together. This is currently needed as DNA sequencing technology cannot read whole genomes in one go, but rather can read small pieces of between 20 and 30,000 bases, depending on the technology used. Typically, the short fragments, called reads, result from shotgun (random) sequencing of genomic DNA.
De novo sequence assemblers are a type of program that assembles short nucleotide sequences into longer ones without the use of a reference genome. These are commonly used in bioinformatic studies to assemble genomes or transcriptomes.
What follows is a tutorial showing how to submit reads for assembly and selecting parameters for the assembly algorithm.
The BV-BRC Viral Genome Assembly service uses an open source, third-part bioinformatics program- IRMA developed by the CDC. IRMA provides a robust next-generation sequencing assembly solution that is adapted to the needs and characteristics of viral genomes. More information about IRMA (https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-016-3030-6) can be found here IRMA and https://wonder.cdc.gov/amd/flu/irma/irma.html pages.
Tip: See Also: Viral Genome Assembly Service
Using the Viral Genome Assembly Service¶
2. Using the Viral Genome Assembly Service
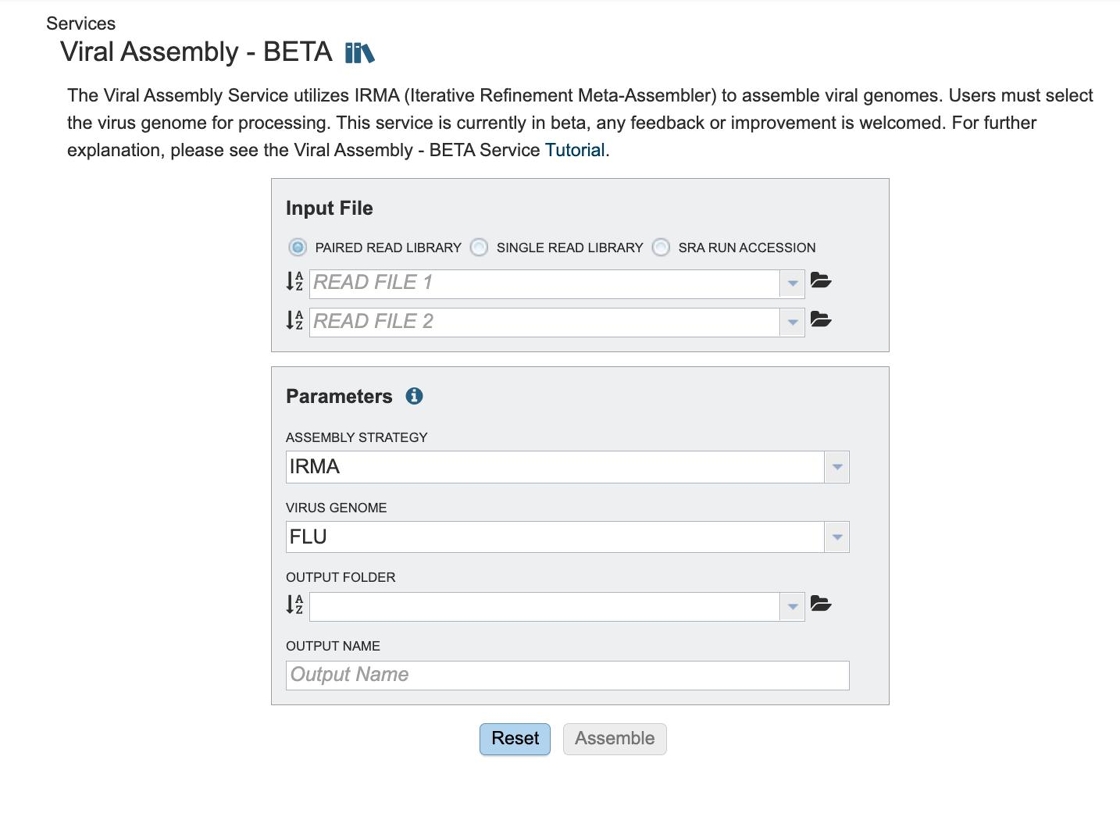
The Viral Assembly submenu option under the Services main menu (Viral Tools category) opens the Viral Genome Assembly input form, shown below. Note: You must be logged into BV-BRC to use this service.
3. Navigate to https://www.bv-brc.org/
4. Click “Viral Assembly”

5. Three options for file input types.
Paired read library
Read File 1 & 2: Many paired read libraries are given as file pairs, with each file containing half of each read pair. Paired read files are expected to be sorted such that each read in a pair occurs in the same Nth position as its mate in their respective files. These files are specified as READ FILE 1 and READ FILE 2. For a given file pair, the selection of which file is READ 1 and which is READ 2 does not matter.
Single read library¶
Read File: The fastq file containing the reads.
SRA run accession¶
Allows direct upload of read files from the NCBI Sequence Read Archive to the BV-BRC Assembly Service. Entering the SRR accession number and clicking the arrow will add the file to the selected libraries box for use in the assembly.
Files accepted by BV-BRC Assembly Service¶
The assembly service accepts read files in either fastq, fasta, fastq.gz, or fasta.gz format.
FASTQ is a text-based format for storing both a nucleotide sequence and its corresponding quality scores. Both the sequence letter and quality score are each encoded with a single ASCII character. A FASTQ file normally uses four lines per sequence:
Line 1 begins with a ‘@’ character and is followed by a sequence identifier and an optional description (like a FASTA title line).
Line 2 is the raw sequence letters.
Line 3 begins with a ‘+’ character and is optionally followed by the same sequence identifier (and any description) again.
Line 4 encodes the quality values for the sequence in Line 2 and must contain the same number of symbols as letters in the sequence.
FASTA is a text-based format for representing either nucleotide sequences or amino acid (protein) sequences using single-letter codes. The format allows for sequence names and comments to precede the sequences. A FASTA read file has two parts:
Line 1 begins with a ‘>’. Everything from the beginning ‘>’ to the first whitespace is considered the sequence identifier. Everything after that is considered the sequence description (this can be metadata, machine serial number, read orientation, etc.), all in a single line.
Line 2 has the sequence, which can span multiple lines depending on the length.

6. Click the “Sra Run Accession” field.

7. You can see the parameters for this service here.
Parameters¶
Assembly Strategy:
IRMA - IRMA is currently the only viral assembly tool. Any other tool can be recommended or requested.
Setting Parameters¶
Currently the only assembly strategy available is IRMA.

8. Click “FLU”. The Flu module has been tested thoroughly the others are still in testing stages. Feel free to add feedback as necessary.
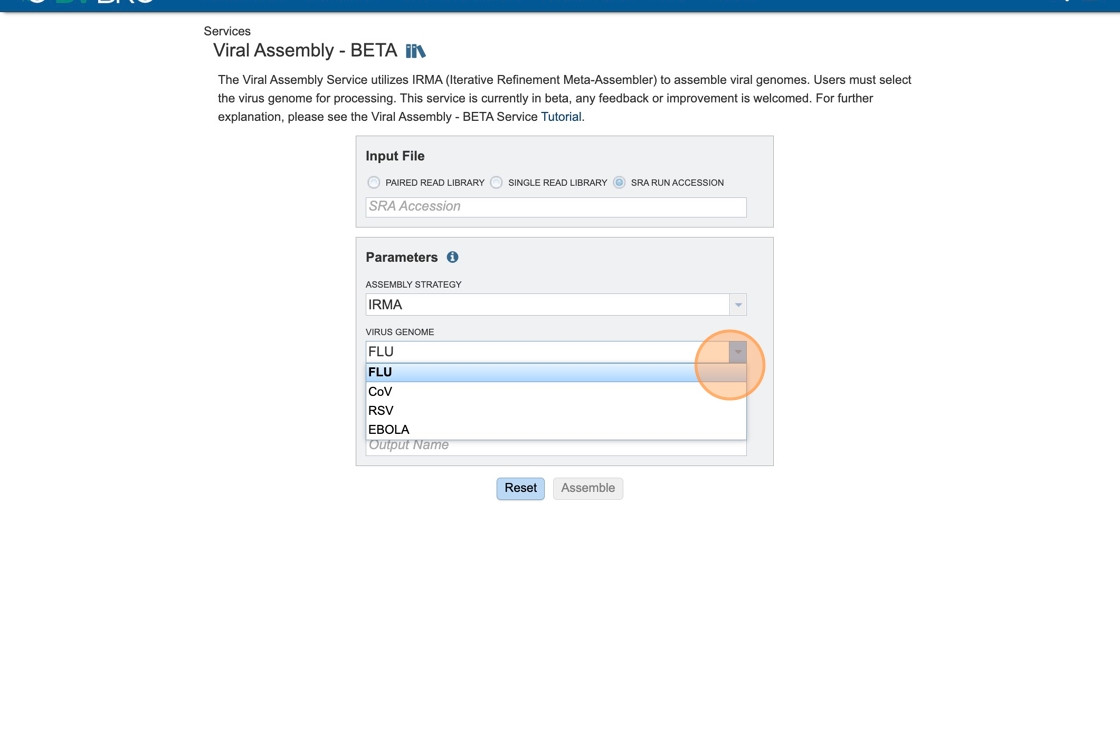
9. An output folder must be selected for the assembly job. Typing the name of the folder in the text box underneath the words Output Folder will show a drop-down box that shows close hits to the name, and clicking on the arrow at the end of the box will open a drop-down box that shows the most recently created folders. To find a previously created folder, or to create a new one, click on the Folder icon at the end of the text box. This will open a pop-up window that shows all the previously created folders.

10. Put your job in whatever folder you feel like having it in.

11. Name the job whatever you want it to be called- for the example it is Assembly_workshop_prep.

12. Click “Tutorial” to view this tutorial if you have any questions.

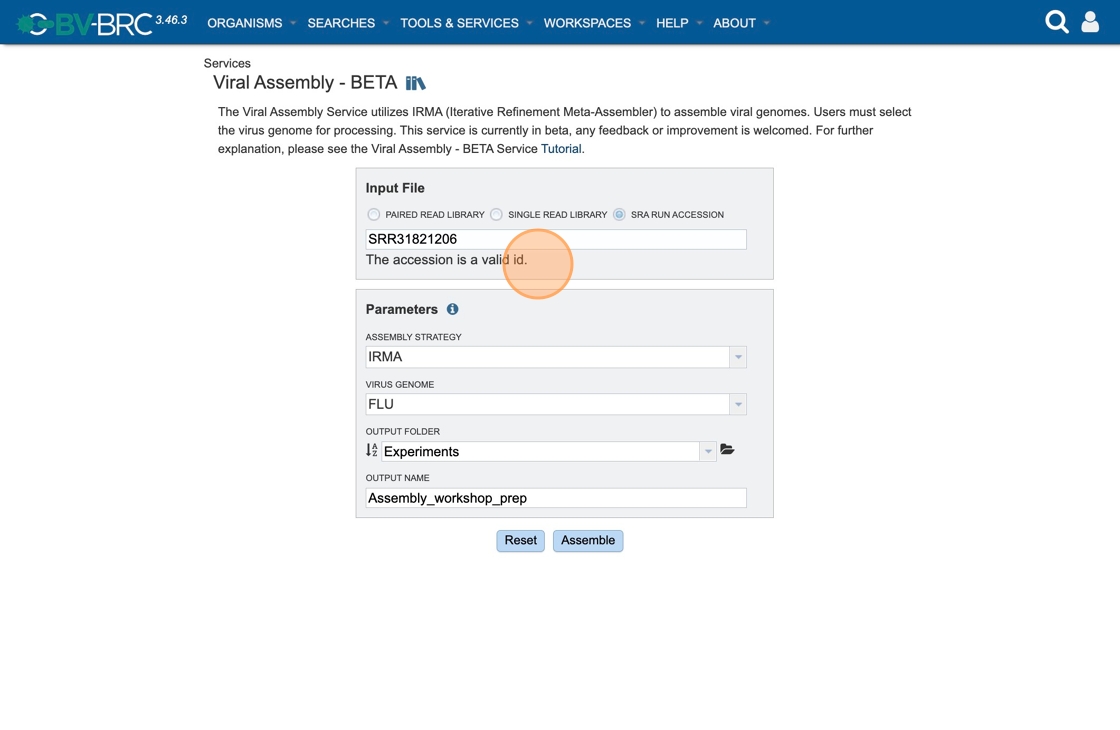
13. Type in the SRA accession number of interest. For the example: SRR31821206

14. Click “Assemble”

15. Check that “The accession is a valid id.”
16. Click “Submitting Assembly Job”

17. A message will appear at the bottom of the page, indicating that the submitted job has entered the BV-BRC queue.

18. Clicking on the Jobs box at the bottom right of any BV-BRC page/

19. ## Viewing and Interpreting the Assembly Job Results
On the jobs page, click on the row that has the assembly of interest. This will populate the vertical green bar on the right with possible downstream steps, which include viewing the results of the job, or reporting an issue that was experienced (like a job failure). Click on the View icon.
Double-click your completed job to view result.

20. This will rewrite the page to show the information about the assembly job, and all of the files that are produced when the pipeline runs.
The information about the job submission can be seen in the table at the top of the results page. To see all the parameters that were selected when the job was submitted, click on the Parameters row. This will show the information on what was selected when the job was originally submitted.
You can click on the output files of the job to see the .fasta
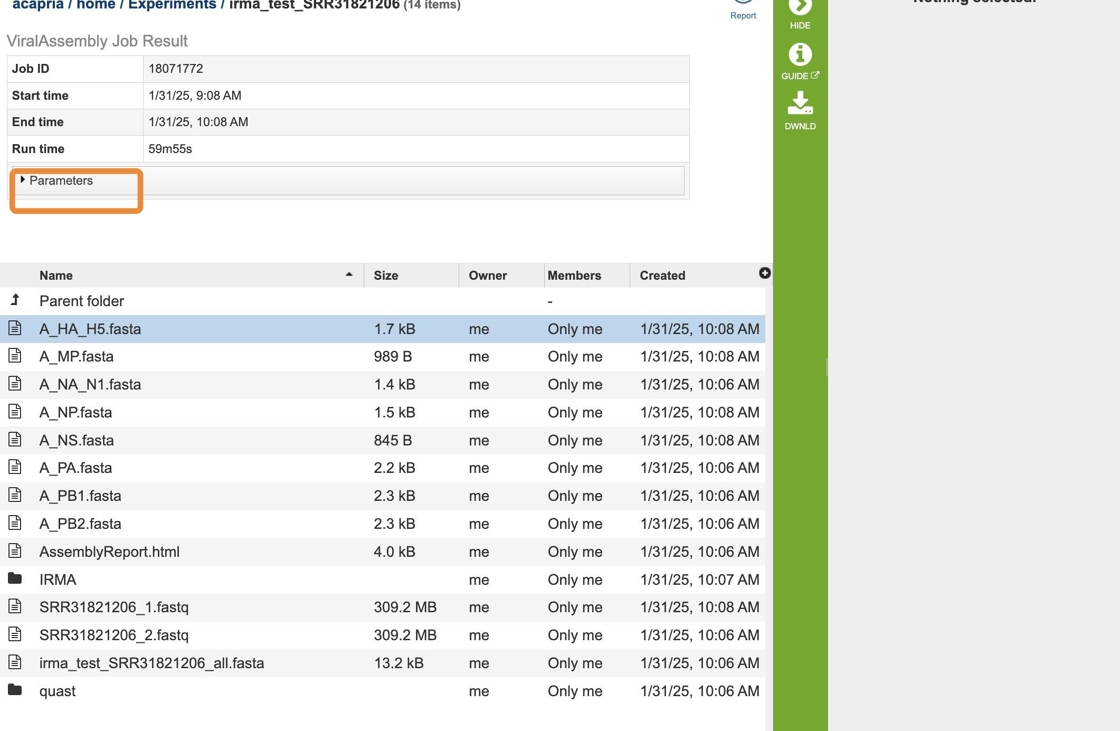
21. You can click on the report icon to see the report of this job.

22. The Genome Assembly report contains valuable information about the assembly, including the number of contigs. Clicking on the row that contains the number of contigs, depth and coverage. AssemblyReport.html will highlight it in blue and populate the action bar with possible downstream steps. Click on the View icon.

23. You can click on the output files of the job to see the .fasta
Click “A_MP.fasta” Or any of the fasta files to get the different fasta’s for each segment. You can download with the download icon on the green bar.
The whole point of the assembly service is the assembly of a contig file from the submitted reads. The contig files can be used in downstream services. Note that the file, which can be clicked on from the Jobs page, has the type matched as “contigs” in the information panel beyond the green bar. The contig file can be used as is in BV-BRC or downloaded for use in other resources or pipelines.

24. Double-click “IRMA” to view the IRMA output files.

25. Double-click “amended_consensus” to see into that folder. Any of this can be used for further analysis.

26. Click “IRMA”
27. Click “.irma_test_SRR31821206”
28. Click this arrow to go back to the directory above as needed.
29. Double-click “irma_test_SRR31821206” This is an alternative way to get to the job run. The job is marked by a flag icon.

30. Click “All statistics are based on contigs of size >= 300 bp, unless otherwise noted (e.g., “# contigs (>= 0 bp)” and “Total length (>= 0 bp)” include …”
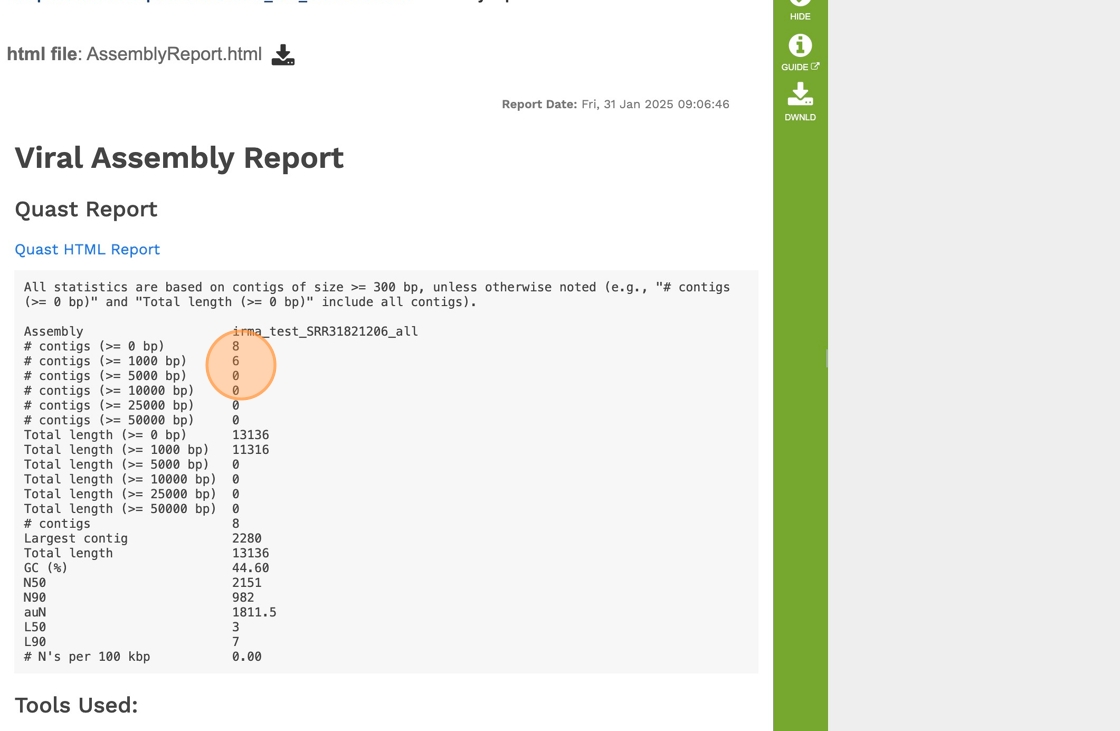
31. Click “Quast HTML Report”
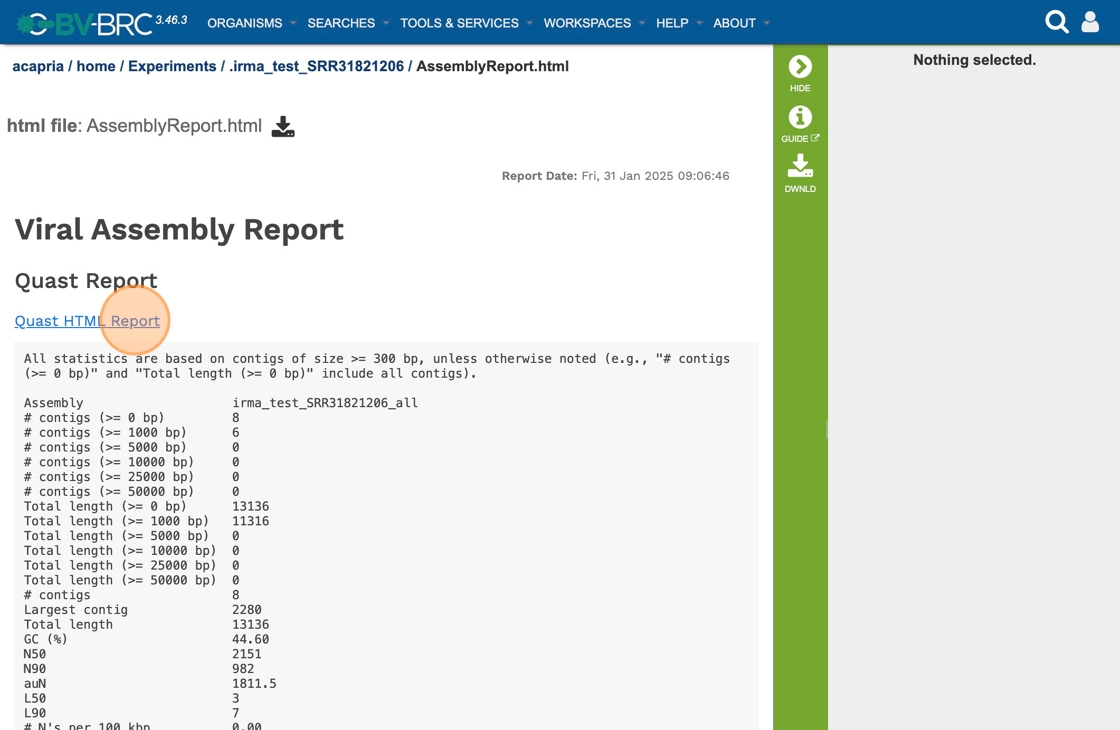
32. Scrolling down will reveal a Quast report, which is also included in the Genome Assembly report. Quast is a quality assessment tool for evaluating and comparing genome assemblies, and shows statistics of the contigs generated in the assembly.
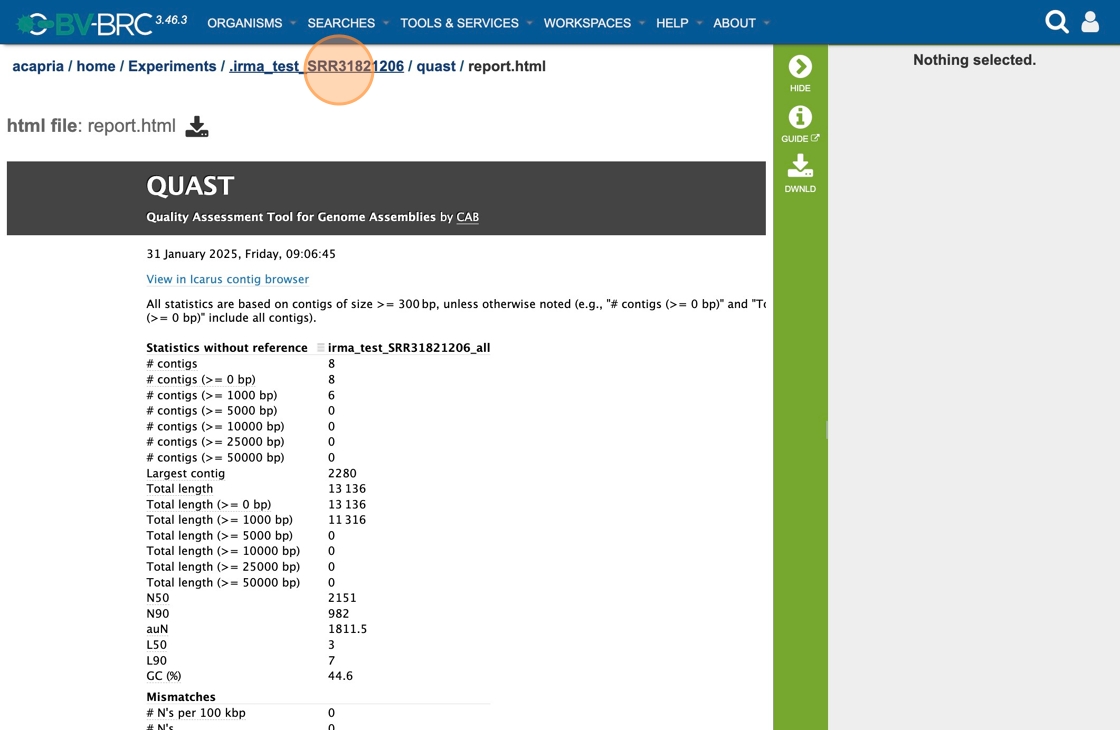
33. Double-click “quast”

34. Click “GC_content_plot.pdf” You can download the GC content plot for further analysis.
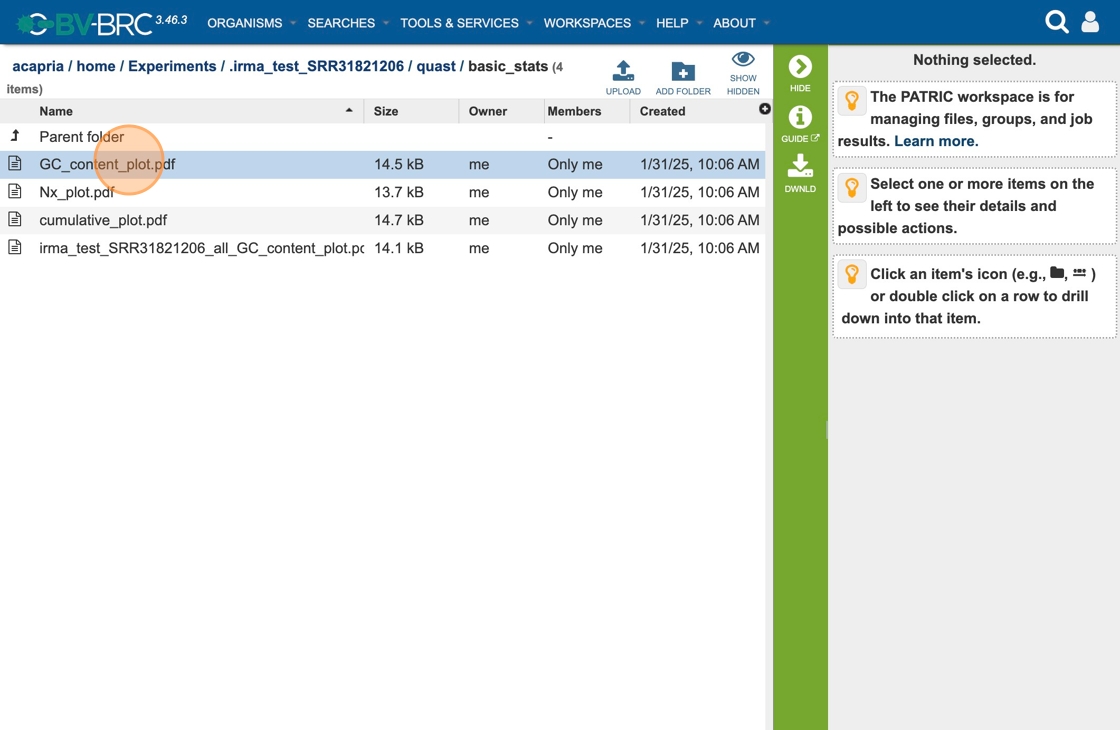
35. The Viral Assembly Service generates several files that are deposited in the Private Workspace in the designated Output Folder. These include
assembly_report.html - Web-viewable report of the assembly including information about the submitted reads and assembly process used.
quast - Folder including all of the standard outputs of quast.
IRMA - Folder including the VCF, bam and bam.bai files generated by IRMA. It also includes sub folders for amended)consensus, figures, intermediate, log, matrices, secondary and tables.
.fasta - All of the associated fasta files for the assembly, including by segment and cummulative.