Metadata-driven Comparative Analysis Tool (meta-CATS)¶
Revised: February 22, 2024
Overview¶
The meta-CATS metadata genome comparison tool takes sequence data and determines the aligned positions that significantly differ between two (or more) user-specified groups. Once an analysis is started, a multiple sequence alignment is performed if the input was unaligned (such as from a database query). A chi-square test of independence is then performed on each non-conserved column of the alignment, to identify those that have a non-random distribution of bases. A quantitative statistical value of variation is computed for all positions. Columns that are perfectly conserved will not be identified as statistically significant. All other non-conserved columns will be evaluated to determine whether the p-value is lower than the specified threshold value. Terminal gaps flanking the aligned sequences will not be taken into account for the analysis.
See also¶
Using the Meta-CATS Service¶
The Meta-CATS submenu option under the “SERVICES” main menu (Viral Services category) opens the Meta-CATS Service input form. Note: You must be logged into BV-BRC to use this service.

Below is a screenshot of the Meta-CATS landing page, as well as a summary of customizable parameters.
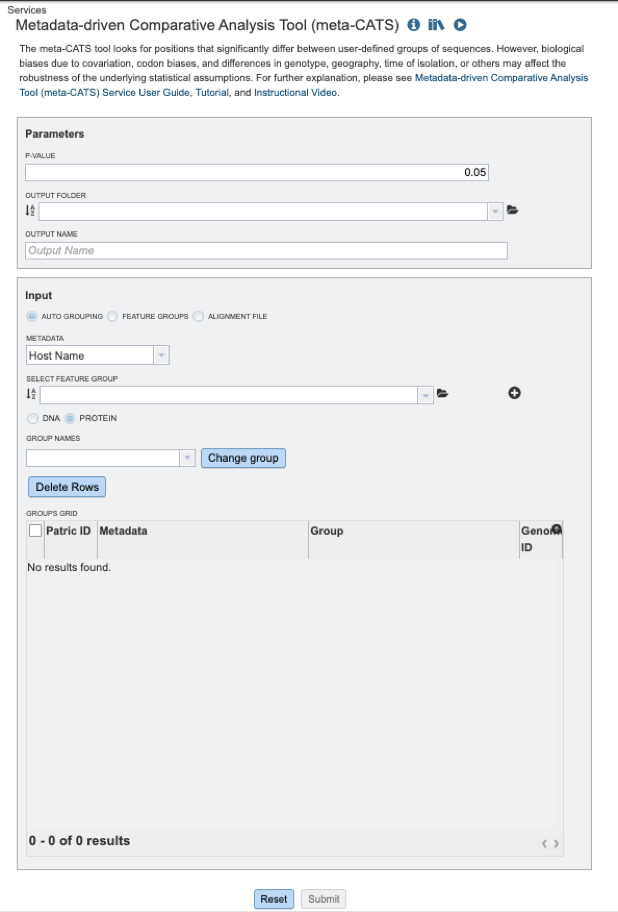
Parameters¶
P-value: the probability of the observed data given that the null hypothesis is true.
Output Folder: The workspace folder where results will be placed.
Output Name: A user-specified label. This name will appear in the workspace when the analysis job is complete.
Fasta Text Input: Users may enter custom sequences here by pasting in FASTA formatted sequences.
Input Options¶
Auto Grouping: Allows users to group sequences by available metadata such as: host, country, year, virus type, host age, etc. The appropriate metadata field may be selected from the “METADATA” drop-down menu.
And/or Select Feature Group: Users may input a nucleic acid or protein FASTA file containing a previously selected “Feature Group” (eg. CDS, tRNA etc.) from their workspace here, either in addition to the FASTA text input, or as an alternative.
Metadata: Auto grouping by available metadata options includes: Host name, geographic location, isolation country, species, genus, and collection year.
Feature Groups: This option allows users to select previously identified groups of sequences saved to their workspace.
Alignment File: This option allows users to select a previously aligned group of nucleotides or proteins.
Select Feature Group: This option allows users to specify feature groups previously saved to their workbench.
DNA/PROTEIN: Allows users to specify whether their sequences are nucleic acid or protein sequences.
Group names: User-specified names for custom groups.
Delete Rows: Allows users to delete unwanted sequences.
Buttons¶
Reset: Resets the input form to default values
Submit: Launches the MSA job. A message will appear below the box to indicate that the job is now in the queue.
Output Results¶
Clicking on the Jobs indicator at the bottom of the BV-BRC page open the Jobs Status page that displays all current and previous service jobs and their status.
Once the job has completed, selecting the job by clicking on it and clicking the “View” button on the green vertical Action Bar on the right-hand side of the page displays the results files (red box).
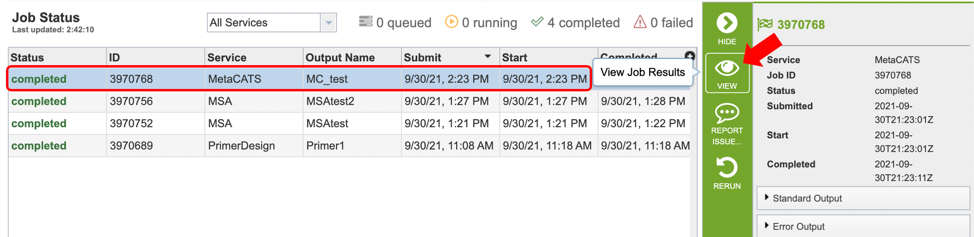
The results page will consist of a header describing the job and a list of output files, as shown below.
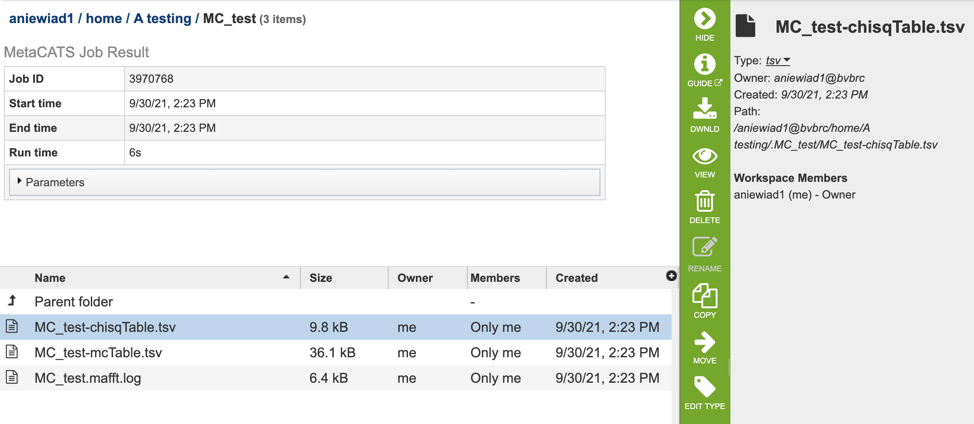
The Meta-CATS Service generates several files that are deposited in the Private Workspace in the designated Output Folder. These include:
chisqTable.tsv - a tab separated value file with results for a “Chi-square Goodness” of fit test result: ie: positions that have significant non-random distribution between the specified groups
mcTable.tsv - a tab separated value file with results for adjusted p-values for multiple comparisons.
Mafft.log - an output log file produced by the Mafft aligner.